Hola de nuevo a todos aquellos lectores habituales, lectores ocasionales, científicos, aficionados y demás indignados y personajillos en general que de vez en cuando, y a pesar de la que está cayendo, decidís pasaros por este 2.0 mini sitio para ver lo que escribe este cochambroso personajillo que soy yo.

El artífice principal de esta loca idea de poner a la gente a doblar proteínas cual negritos cogiendo café, fue del genial y jovencísimo investigador David Baker.




Debería de comenzar disculpándome por haber tardado tanto en escribir, pero creedme que he estado escribiendo y mucho. Quizá debería deciros que lo siento mucho….que no volverá a pasar… que no era mi intención dejaros abandonados… y mientras, las visitas casi llegan a 10.000. De modo que no me queda otra que dar las gracias y volver a nuestros orígenes “blogueriles”.
Efectivamente, ¿Os acordáis de aquellas proteínas que se hacían la permanente? En este enlace podéis ver la primera entrada de este blog, donde hablábamos de las IDP´s (Intrinsically Disrodered Proteins) y de cómo hacemos en ciencia para ver lo invisible. ¿Y de aquella otra entrada que hablaba sobre la dificultad de predecir la estructura de una proteína y la de movimientos que puede hacer?, y por último quiero recordaros esta otra entrada, donde con un título un tanto soez, os hablaba de la iniciativa CASP.
Pues bien, en esta ocasión vamos a hablar de uno de los métodos de predicción de estructura de proteínas que más éxito tiene y que mejores resultados está dando. (A pesar de que como todo tiene sus inconvenientes).
Existen básicamente 3 métodos o estrategias que podemos utilizar para hacer la predicción de la estructura de una proteína:
1 – Métodos estadísticos o de Fold recognition o Threading. Se trata de métodos basados en la propensión de las secuencias a ciertas estructuras secundarias (en láminas, en bucles, espirales, etc), las características fisico-químicas de los aminoácidos que forman la proteína, la accesibilidad al disolvente, etc. Es decir, son cuestión de pura estadística y lógica físico-química.
2 – Métodos Ab inicio - o métodos de predicción de Novo. Se trata de métodos que pretenden hacer una predicción 3D de una estructura de una proteína, basándose exclusivamente en la secuencia de aminoácidos y sin usar las estructuras de proteínas ya conocidas o predeterminadas.
3 – Métodos por Homología. Son aquellos que se basan en un parecido significativo de secuencias con proteínas de las que ya se conoce la estructura, para predecir la nueva estructura. Desde luego son los más recomendables siempre y cuando tengamos una proteína que se parezca lo suficiente a la nuestra y tengamos su estructura.
Pero, ¿Porqué nos empeñamos en predecir estructuras en lugar de medirlas?, ¿Acaso los científicos se han pasado al mundo de la farándula y ahora usan bolitas de cristal mágicas en lugar de difractares de rayos X? Pues la verdad es que no… aunque no estaría del todo mal ¡Quizás puede ser una forma de sacar dinero para la ciencia ahora que no tenemos ni ministerio asignado!
Intentamos predecir las estructuras, porque como ya mencioné en su momento, purificar, cristalizar y determinar la estructura de una proteína no es nada fácil. Además, si conocemos las reglas del juego (como se doblan las proteínas) podremos diseñar in silico proteínas con nuevas funciones o mejorar las que ya conocemos. De momento hay una cosa clara. Una función, viene determinada por una estructura, y una estructura determinada se puede construir con una secuencia de aminoácidos dada…..pero mucho me temo que al revés no es tan fácil.
Hace relativamente poco tiempo aparecieron en el periódico estás noticias:
Tras estos sensacionalistas y agoreros titulares hay toda una base o procedimiento que intenta acercarse a entender las reglas del juego (valga la ironía) de las proteínas. Se trata de un videojuego, si, un videojuego. Solo que en este caso al jugar, se hace ciencia de verdad, de la que se publica en revistas científicas. Se llama "Foldit" , algo así como “Dóblalo” o “Pliégalo”.
Foldit es un juego de ordenador experimental que consiste básicamente en jugar a mover una proteína hasta que alcance un nivel de mínima energía, es decir, intentar predecir la estructura tridimensional de la proteína atendiendo a su plegamiento en base solo a la secuencia de aminoácidos conocida. Su objetivo es encontrar, gracias en parte a la intuición y en graaaaaan parte a la suerte del jugador, las formas naturales de las proteínas al plegarse.

Se trata de un juego basado en la plataforma Rosetta, fruto de la colaboración del departamento de Bioquímica y el de informática e ingeniería de la universidad de Washington, en el que básicamente se sirven de pequeñas e ineficaces computadoras (vuestros cerebros) capaces de cometer fallos y de probar las cosas más inverosímiles en modelado de proteínas (¡¡¡porque como lo puede usar cualquiera…!!!) para, en parte por intuición y en parte por suerte, probar todas las formas en las que un fragmento de proteína se puede doblar hasta alcanzar es de máxima estabilidad y, por tanto, mínima energía. SI, OS UTILIZAN DE GRATIS… PERO EL JUEGO MOLA.
Ahí esta el tío, tan entretenido en doblar que ni de peinarse tiene tiempo. ¡APRENDED MALDITOS VAGOS!.
David Baker es un bioquímico y biólogo computacional Americano, nacido en Seattle en 1962 que tuvo una fantástica idea a la que denominó Rosetta (en honor a la Piedra Rosetta, un fragmento de una antigua estela egipcia de granodiorita inscrita con un decreto escrito en 3 lenguas en el año 196 a .C. en nombre del faraón Prolomeo V. Al estar escrita en jeroglífico egipcio, en escritura demótica y griego antiguo, ayudó a descifrar y entender gran parte de las culturas que, como la egipcia, no tienen un alfabeto como tal).
La idea Rosetta es muy sencilla.

- Cogemos una secuencia de una proteína cualquiera de la que queramos saber su estructura y la partimos en fragmentos de 9 aminoácidos al azar.
- Después a cada uno de esos fragmentos se le busca una conformación, utilizando simulaciones de función de energía (como cuando doblas un alambre, hay formas que son estables y otras que no se mantienen por más que insistamos).
- Con todas esas mini estructuras, se recurre a una librería de fragmentos donde comparamos con estructuras de proteínas ya conocidas para estimar la probabilidad de que ese “doblado” sea el más adecuado.
- Y por último, se unen los fragmentos en varios órdenes y se estima como queda la proteína en base a la estabilidad energética de la estructura total etc.
A continuación, se estima……ESPERA….UN MOMENTO……¿¿¿¿SE RECURRE A UNA LIBRERÍA DE FRAGMENTOS DE ESTRUCTURAS YA CONOCIDAS????, ¡¡¡PUES VAYA TIMO!!!, ¿No se suponía que era un método Ab initio? Bueno, ya os dije que no era perfecto, pero funciona (a veces).
Este proceso que os he comentado, se repite unas 1000 veces cortando la proteína en diferentes sitios, haciendo diferentes foldings, poniendo las mini estructuras de 9 aminoácidos en diferentes órdenes, etc. Pues bien, el juego de Foldit, es básicamente, determinar a mano (o más bien a ojo) la estructura de esos fragmentos de 9 aminoácidos y mandarlas a una base de datos que las procesa y ordena. Os recomiendo que os deis una vuelta por la página del tal David, y leáis algunas de las cosas que se dicen de él "Enlace".
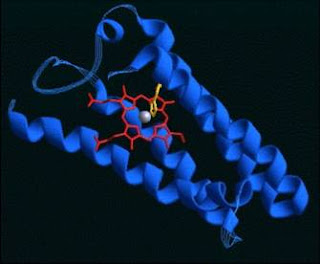
Pues no os lo vais a creer, pero el método funciona. Por procesos muy parecidos al de Rosetta, otro servidor, en esta ocasión, Scratch/3Dpro, de la Universidad de California, se diseñó una proteína artificial, no existente en la naturaleza. Se trata de una proteína muy sencilla, consistente en 4 hélices alpha en cuyo centro acomplejan un tetrapirrol que contiene hierro (los más avispados ya habrán deducido que puede servir para transportar o almacenar oxígeno). Cada una de las hélices está formada por tan solo 27 residuos (ELLKLLEELLKKLEELLKLLEELLKKL) y la proteína realmente sirve para transportar oxígeno (aunque la hemoglobina de nuestros glóbulos rojos lo sigue haciendo mucho mejor).
Por ponerle algún ¡pero!, Scratch solo hace predicción de estructuras teniendo en cuenta los Carbonos alpha…nada más. Lo cual no da mucha información de las cadenas laterales de los aminoácidos ni la interacción entre ellas.
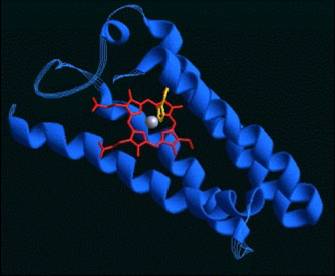

Lo que comenzó siendo una llamada para que los voluntarios dejaran que los sistemas informáticos que se dedican a estudiar el plegamiento de proteínas les prestaran la capacidad de sus ordenadores, paso a ser un juego interactivo con los jugadores diseñando esas estructuras. Pero se ha ido más allá y se han ido ensamblando puzzles hasta conseguir moléculas artificiales hasta 20 veces más eficaces que las naturales (aunque yo siempre digo que eso no es difícil, la naturaleza está diseñada para vivir, no optimizada con lógica y matemática). Los resultados han sido publicados en Nature Biotechnology. Es lo que algunos ya han venido a llamar la ciencia de la intuición.
Sinceramente, os recomiendo que juguéis, es entretenido. Pero no esperéis gran cosa. Para mi se parece mucho a darle las piezas de un puzzle a miles de monos y esperar que vayan montando trozos para yo luego llegar y unirlos con lógica… no hacen falta más comentarios. Pero sin duda el sistema funciona y ha sido una verdadera revolución.
D. Baker ya ha propuesto el siguiente reto… fabricar inhibidores más potentes para curar el virus de la gripe. ¿Medicamentos a la vista?
PD: No quiero olvidarme de recordar a un gran químico que nació tal día como hoy de hace 100 años. Se trata de Glenn Theodore Seaborg (19.04.1912 – 25.02.1999).
Fue premio Nobel de química en 1951 por sus trabajos en la química de los elementos transuránicos, contribuyendo al descubrimiento y aislamiento de diez elementos químicos, desarrolló el concepto de elemento actínido y fue el primero en proponer la serie actínida que tantos quebraderos de cabeza me dio en bachillerato. Se le puede considerar un pionero de la química nuclear contribuyendo al descubrimiento del hierro 59 entre otros cientos de isótopos. Este hierro 59 fue especialmente útil par el estudio de la hemoglobina en la sangre humana. En 1938, junto a su colaborador Livingood, crearon el isótopo de yodo, el yodo-131 que aún a día de hoy se emplea para el tratamiento de la glándula tiroides.
Seaborg es un gran personaje dentro de la química, uno de tantos trabajadores incansables de la ciencia que han hecho avanzar nuestra sociedad y sobretodo la medicina. Pero además consiguió algo con lo que muchos hemos soñado alguna vez, "Prolongar la vida de su propia madre con sus descubrimientos". El tratamiento con Yodo-131 consiguió prolongar la vida de su madre y eso le valió a Seaborg ser aclamado como uno de los pioneros en la aplicación de medicina nuclear, base de los actuales radiofármacos o radiotrazadores que tanto ayudan al diagnóstico y tratamiento de muchas enfermedades.
Esta entrada participa en la XIV Edición del Carnaval de Química que organiza en esta ocasión Bernardo Herradón desde Educación Química. También participa en elXII Carnaval de Biología que organiza Raúl de la Puente en Blog de Laboratorio.
ATG
ResponderEliminarMuy buena entrada Óscar, qué recuerdos de esas primeras entradas jaja. Un detalle por cierto el recuerdo a Seaborg.
Mucho me gustaría que lográsemos predecir estructuras proteicas a partir de solo secuencia aminoacídica sin esas "librerías de estructuras conocidas" y demás.. ¡ si solo conociéramos todas las variables que intervienen en el proceso ! Y lo más seguro es que si se supiesen las variables en unas condiciones, probablemente serían también variables entre condiciones u organismos. No obstante no dudo que se conseguirá algún día.
Y mientras tanto, pues nada, cuando haya ratos libres, a foldinear un poco jajaja
TAA